 2017, Vol. 19
2017, Vol. 19



2. 复旦大学儿科研究所, 上海 201102
患儿,男,2月余。因反复纳差伴皮肤色素沉着2月余入院。患儿生后22天时因皮肤黄染伴纳差住院,查体发现皮肤暗黄;血常规白细胞12.4×109/L,中性粒细胞比例33.1%、淋巴细胞比例59.8%,余项正常;C反应蛋白正常;血钾8.0 mmol/L(参考值3.5~5.5 mmol/L),血钠125 mmol/L(参考值135~145 mmol/L),血钙、氯正常;肝功能:谷丙转氨酶(ALT)65 IU/L(参考值0~40 IU/L),谷草转氨酶(AST)76 IU/L(参考值0~40 IU/L),总胆红素(TB)175 μmol/L(参考值5~17 μmol/L)、直接胆红素(DB)139 μmol/L(参考值0~6 μmol/L);肾功能无异常;肌酸激酶(CK):19 949 IU/L(参考值25~200 IU/L),肌酸肌酶同工酶(CK-MB):386 IU/L(参考值0~25 IU/ L);甲状腺功能:总T3、T4及游离T3、T4均正常,TSH 11.28 μIU/mL(参考值0.25~7.31 μIU/mL);促肾上腺皮质激素(ACTH)1 250 pg/mL(参考值0~46 pg/mL);血皮质醇5 μg/dL(参考值5~ 25 μg/dL),睾酮160 ng/mL(参考值0~32 ng/mL);17α-羟孕酮正常;肾素0.01 ng/mL(参考值0.13~1.94 ng/mL)。甲肝、乙肝、丙肝抗体均阴性。TORCH阴性。头颅MRI平扫未见明显异常。腹部B超:肝肿大,空腹胆囊不充盈,总胆管未见扩张,脾、胰、双肾未见局灶性占位;双侧肾上腺探测不清。诊断为婴儿肝炎综合征、先天性肾上腺皮质增生症、暂时性甲状腺功能低下等,予纠正电解质紊乱,护肝利胆以及口服氢化可的松(每次2 mg,Q8 h)、优甲乐(25 μg/d)等治疗。治疗2天后复查血电解质正常。2周后复查ALT 122 IU/L,TB 199.4 μmol/L,DB 176.8 μmol/L,较治疗前均有增高;TSH恢复正常;ACTH 1 250 pg/mL,皮质醇4 μg/dL(参考值5~25 μg/dL),睾酮降至107 ng/mL。出院后继续服用氢化可的松、优甲乐,吃奶稍差,哭声低弱。因纳差加重,皮肤色深无明显改善,再次入院。
患儿系第二胎第一产,出生胎龄41周,剖宫产出生,出生体重4 000 g,出生时无窒息、1 min Apgar评分10分,生后混合喂养。患儿父母体健,三代家系中无类似疾病患者。
入院体查:呼吸46次/min,心率130次/min,BP 74/44 mm Hg。神志清,营养不良貌,哭声低弱;皮肤色素沉着、粗糙、弹性差,无出血点;四肢稍凉,毛细血管充盈时间 < 3 s。心音有力,心律齐,未闻及杂音。无吸凹征,双肺呼吸音粗,未及明显罗音。腹平软,肝肋下1.5 cm、质软,脾肋下未及。外生殖器无畸形、颜色深黑。双上肢肌张力稍高,双下肢肌张力正常,双下肢无水肿。
实验室检查:血气分析(静脉血)pH 7.25,BE -4.4 mmol/L、PCO2 24 mm Hg(参考值35~45 mm Hg)、PO2正常;尿常规:尿糖+++,酮体+,红细胞4~6/HP,白细胞30~45/HP,蛋白(-),尿胆原(-);血电解质:钾6.4 mmol/L(参考值3.5~5.5 mmol/L)、钠110 mmol/L(参考值135~145 mmo/L),血钙、氯均正常,肝功能ALT 504 IU/L(参考值0~40 IU/L),AST 516 IU/L(参考值0~40 IU/L),TB 169.4 μmol/L(参考值5.1~17.1 μmol/L),DB 154.7 μmol/L;肾功能BUN 7.4 mmol/L(参考值2.5~6.5 mmol/L),肌酐正常;血脂分析:总胆固醇正常,甘油三脂(TG)6.2 mmol/L(参考值0.6~1.7 mmol/L);血常规:白细胞15.6×109/L,红细胞1.56×1012/L,淋巴细胞百分率50.3%,中性细胞百分比37.6%,血红蛋白49.2 g/L,血小板正常。血串联质谱分析:氨基酸谱和酰基肉碱谱无明显异常。尿气相色谱质谱分析:2-酮戊二酸升高,其他无明显异常。
2 诊断思维患儿新生儿期出现皮肤黄染及色素沉着,实验室检查发现肝功能损害(直接胆红素增高为主的高胆红素血症及转氨酶进行性增高),肌酸激酶高达19 949 IU/L,ACTH高达1 250 pg/mL,以及甘油三脂增高,皮质醇降低。感染或胆道畸形可导致转氨酶增高和直接胆红素增高为主的黄疸,但患儿TORCH阴性、病毒性肝炎抗体阴性,无常见病毒感染的依据,超声检查也不支持胆道畸形。患儿为小婴儿,肌酸激酶显著增高,需注意杜氏肌营养不良(Duchenne muscular dystrophy, DMD)、线粒体肌病等遗传代谢性疾病。但这些疾病均不能解释同时合并的ACTH明显升高、皮质醇水平降低以及婴儿期出现的高甘油三脂血症。ACTH升高伴皮质醇水平降低的常见原因为先天性肾上腺发育不良(adrenal hypoplasia congenital, AHC)或者先天性肾上腺皮质增生症(congenital adrenal hyperplasia, CAH)。而且肾上腺皮质功能不全也可导致肝功能损害,虽然比较少见[1]。ACTH激发试验有助于AHC和CAH的鉴别。静脉推注ACTH,如果皮质醇无明显升高,睾酮、孕酮、17α-羟孕酮等激素水平也无明显变化,提示AHC,基因分析可助确诊。CAH是一类常染色体隐性遗传病,常见的基因突变为CYP21、CYP11、CYP17等;而AHC为一种X连锁隐性遗传病,是由位于Xp21的编码核转录因子DAX-1等基因突变引起。该患儿婴儿期即出现高甘油三脂血症,需要注意先天性脂质代谢紊乱和脂肪酸能量代谢障碍,但先天性脂质代谢紊乱早期即引起肝损和肌酶显著增高未见报道;脂肪酸能量障碍可引起低血糖、肉碱和酰基肉碱水平异常等情况,但肌酶增高仅为轻中度,而且患儿血糖正常,无肉碱和酰基肉碱水平异常。因此没有某种单一疾病能解释该患儿的所有特点,需要注意某些少见综合征,进行基因芯片检测有助诊断。
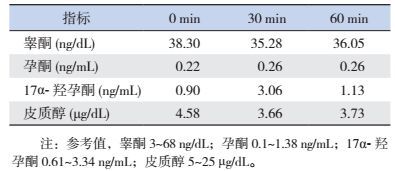
3 进一步检查(1)ACTH激发试验:静脉推注ACTH后,皮质醇无明显升高,睾酮、孕酮、17羟孕酮等激素水平也无明显变化,见表 1。提示肾上腺皮质功能不全。
| 表 1 ACTH激发试验 |
(2)微阵列比较基因组杂交技术(array CGH)检测:对患儿及其父母进行基因芯片检测,发现患儿X染色体短臂(Xp21.3-p21.1)8.7 Mb致病性拷贝缺失(图 1);患儿母亲在相同区域也存在相同片段的拷贝缺失,其父的拷贝数正常。

|
图 1 患儿X染色体aCGH检测示意图 红色区域代表chrX: 28, 612, 178-37, 392, 685部分区段信号降低,提示为微缺失。 |
4 诊断及其依据
患儿确诊为复合型甘油激酶缺乏症(complex glycerol kinase deficiency, cGKD),依据:(1)新生儿期出现皮肤黄染及色素沉着;(2)黄疸以直接胆红素增高为主,转氨酶进行性增高,肌酸激酶高达上万,ACTH高达上千,甘油三脂增高,皮质醇降低伴高钾、低钠血症;(3)患儿及其母亲X染色体短臂(Xp21.3-p21.1)8.7 Mb致病性拷贝缺失。
5 临床经过确诊复合型甘油激酶缺乏症后给予氢化可的松(每日10 mg/m2)替代治疗4年余,及大剂量辅酶Q10(每日5~10 mg/kg)联合左卡尼汀(每日100 mg/kg)治疗,并限制动物油等饱和脂肪酸摄入。随访4年至患儿4岁。治疗1周后患儿皮质醇以及电解质即恢复正常,但随访期间CK、ALT、AST以及TG等进行性升高(表 2),且肌力较差(V级左右),智力发育迟缓。
| 表 2 随访期间患儿血清生化标志物的变化 |

6 讨论
甘油激酶缺乏症(GKD)是一种少见的X染色体隐性遗传性代谢缺陷病,可分为单纯型和复合型(cGKD)。cGKD又称Xp21邻近基因缺失综合征,是包含甘油激酶基因位点的Xp21区域不同大小片段基因缺失所致的综合征,包括甘油激酶缺乏所致的高甘油三脂血症、AHC、DMD以及智力发育迟缓。该病最早于1977年由McCabe等[2]报道,兄弟俩(2~5岁)均表现为尿甘油水平升高、生长落后、智力障碍。Guggenheim等[3]对这2名患儿进行随访,发现还存在肾上腺皮质功能不全。Wieringa等[4]及Patil等[5]发现一家族中患病男性存在DMD、AHC以及GKD的基因异常。
McCabe等[6]总结了cGKD不同相邻基因位点缺失导致的常见症状,包括AHC、DMD、阿兰群岛眼病、慢性肉芽肿病、视网膜色素变性以及鸟氨酸氨甲酰基转移酶缺乏等症状,cGKD临床表现取决于所累及的基因片段位置及长度,但以GKD-AHC-DMD的组合最为常见。
李秀珍等[7]报道3例与cGKD症状相似的男性患儿,均于新生儿期发病,均有肾上腺皮质功能低下、高甘油尿症及DMD的症状,但未行基因检测。彭镜等[8]报道1例68天龄男性婴儿,以反复气促、喉鸣、喂养困难、反应低下入院,有高钾、低钠血症及CK显著增高、尿甘油增高,但无皮肤色素沉着,基因检测提示Xp21.1区域微缺失,确诊为Xp21邻近基因缺失综合征(AHC-GKD-DMD)。本例存在肾上腺皮质功能减退、肝功能损害、肌酶显著增高、高脂血症、电解质紊乱等多系统损害特点,患儿及其母亲均有X染色体短臂(Xp21.3-p21.1)8.7 Mb致病性拷贝缺失,符合cGKD诊断标准。刘晖等[9]也报道1例cGKD,有AHC及智力发育迟缓的症状,尿甘油水平也增高,但未行基因检测。王旭等[10]对1例cGKD患者进行半年随访,治疗后CK从8 539.4 U/L降至4 783 U/L。而本例患儿随访4年,CK进行性升高,并有肌力减退和智力发育迟缓,提示对于该病应注意智力及运动功能的长期随访。IL1RAPL1基因位于Xp21相邻区段内,主要在中枢神经系统表达,IL1RAPL1基因缺失是智力发育障碍的病因之一[11-12]。本例cGKD患儿智力落后可能与Xp21.3-p21.1致病性拷贝缺失中包含有IL1RAPL1基因缺失相关。
cGKD的治疗以对症为主,包括氢化可的松替代治疗、低脂饮食以及康复治疗,预后与确诊时间以及基因片段缺失大小相关。
7 结语本病例新生儿期起病,存在肾上腺皮质功能减退、肝功能损害、肌酶显著增高、高脂血症、电解质紊乱等多系统损害,难以用单一疾病解释,最终结合aCGH检测诊断为cGKD。因此对于表现为肾上腺皮质功能不全者,应注意肌酸激酶以及甘油三脂水平,必要时行aCGH检测以及早诊断。
| [1] | Burra P. Liver abnormalities and endocrine diseases[J]. Best Pract Res Clin Gastroenterol, 2013, 27 (4): 553–563. DOI:10.1016/j.bpg.2013.06.014 |
| [2] | McCabe ER, Fennessey PV, Guggenheim MA, et al. Human glycerol kinase deficiency with hyperglycerolemia and glyceroluria[J]. Biochem Biophys Res Commun, 1977, 78 (4): 1327–1333. DOI:10.1016/0006-291X(77)91437-1 |
| [3] | Guggenheim MA, McCabe ER, Roig M, et al. Glycerol kinase deficiency with neuromuscular, skeletal, and adrenal abnormalities[J]. Ann Neurol, 1980, 7 (5): 441–449. DOI:10.1002/(ISSN)1531-8249 |
| [4] | Wieringa B, Hustinx T, Scheres J, et al. Complex glycerol kinase deficiency syndrome explained as X-chromosomal deletion[J]. Clin Genet, 1985, 27 (5): 522–523. |
| [5] | Patil SR, Bartley JA, Murray JC, et al. X-linked glycerol kinase, adrenal hypoplasia and myopathy maps at Xp21[J]. Cytogenet. Cell Genet, 1985, 40 : 720–721. |
| [6] | McCabe ER, Towbin JA, van den Engh G, et al. Xp21 contiguous gene syndromes: deletion quantitation with bivariate flow karyotyping allows mapping of patient breakpoints[J]. Am J Hum Genet, 1992, 51 (6): 1277–1285. |
| [7] | 李秀珍, 刘丽, 梅慧芬, 等. 儿童复合型甘油激酶缺乏症[J]. 中国当代儿科杂志, 2007, 9 (5): 441–444. |
| [8] | 彭镜, 尹飞, 吴丽文, 等. Xp21邻近基因缺失综合征一例及其分子遗传学诊断[J]. 中华儿科杂志, 2009, 47 (10): 792–793. DOI:10.3760/cma.j.issn.0578-1310.2009.10.017 |
| [9] | 刘晖, 王子敬, 郑启安, 等. 复合型甘油激酶缺乏症1例报告[J]. 临床儿科杂志, 2012, 30 (3): 286–290. |
| [10] | 王旭, 吴迪, 方方, 等. Xp21临近基因缺失综合征6例临床和遗传学研究[J]. 中国实用儿科杂志, 2015, 30 (7): 535–539. |
| [11] | Zhang YH, Huang BL, Niakan KK, et al. IL1RAPL1 is associated with mental retardation in patients with complex glycerol kinase deficiency who have deletions extending telomeric of DAX1[J]. Hum Mutat, 2004, 24 (3): 273. |
| [12] | Behnecke A, Hinderhofer K, Bartsch O, et al. Intragenic deletions of IL1RAPL1: Report of two cases and review of the literature[J]. Am J Med Genet A, 2011, 155A (2): 372–379. |