 2017, Vol. 19
2017, Vol. 19



戊二酸尿症1型(glutaric aciduria type 1, GA1)是一种常染色体隐性遗传疾病,该疾病各种族发生率不同,台湾地区统计约1/106 474,国内浙江省统计约1/64 708[1-3]。戊二酰辅酶A脱氢酶(glutaryl-CoA dehydrogenase, GCDH)是色氨酸、赖氨酸、羟赖氨酸代谢途径的关键酶。蛋白质中赖氨酸较色氨酸含量更丰富。GA1病因为编码GCDH的基因发生突变致GCDH活性减低,最终导致赖氨酸代谢障碍,代谢产物戊二酸、3-羟基戊二酸等在体内蓄积而致病[4]。GA1常在患儿6~36个月时因感染、腹泻、手术等非特异性因素诱发急性脑病危象而被发现并诊断。急性脑病危象后遗留不可逆性神经损伤。除脑组织外肝脏GCDH含量亦很丰富,而且肝脏是氨基酸代谢和能量代谢的重要器官。肝脏在GA1发病中占有重要角色,由此推测肝脏亦是GA1受累器官,然而GA1患儿临床表现少有肝脏损伤。既往动物模型研究亦显示脑组织损伤明显强于肝脏[5-8]。研究GA1致神经元和肝细胞损伤的差异性,有助于为寻找GA1治疗的新途径提供新的理论依据。迄今尚无细胞模型研究肝脏的损伤作用。原代肝细胞培养操作过程复杂,培养条件苛刻,慢病毒感染效率难以保证,因此本研究采用正常大鼠永生化的肝细胞BRL-3A细胞(big rat liver-3A cells)进行研究,操作简单,成本低,批间差异小。前期研究已成功构建含shRNA慢病毒载体靶向沉默GCDH基因,慢病毒感染神经元结合高浓度赖氨酸培养成功构建GA1神经损伤细胞研究模型[9]。为了进一步研究GA1对肝脏的损伤作用,本研究构建含shRNA慢病毒载体感染BRL肝细胞系,靶向沉默GCDH基因,结合高浓度赖氨酸培养模拟GA1代谢累积物,通过MTT和Hoechst 33342检测细胞活性及凋亡情况,通过Western blot检测反映细胞凋亡的经典指标Caspase3水平,以进一步评价肝细胞活性。
1 材料与方法 1.1 BRL肝细胞培养液氮中取出冻存细胞(中科院上海细胞库),迅速于37℃水浴中解冻。1 000 r/min离心2 min后超净台中弃上清,新鲜培养基重悬细胞(90%DMEM高糖培养基+10%胎牛血清,均购自美国Gibico公司),调整细胞数约为5×104/孔种植于6孔板。细胞生长至90%融合时胰酶(美国Gibico公司)消化传代。
1.2 慢病毒感染肝细胞及感染率检测细胞融合度约30%时随机分为正常对照组、阴性对照组和GCDH沉默组,正常对照组细胞无任何处理,阴性对照组和GCDH沉默组分别用阴性对照慢病毒和携带靶向沉默GCDH基因的shRNA慢病毒转染(上海吉凯基因化学技术有限公司)[9]。预实验筛选出最佳感染复数(MOI=病毒数/细胞数)为20。慢病毒载体本身携带绿色荧光蛋白,感染72 h后采用荧光显微镜(日本OLYMPUS)观察阴性对照组和GCDH沉默组感染效率。
1.3 Western blot法检测GCDH蛋白表达水平慢病毒感染72 h后弃培养基,PBS液洗细胞2次后,加细胞裂解液收集细胞至Ep管中,50 W超声仪震荡10 s,冰上静置10 min,重复3次。200 mA SDS-PAGE电泳(浓缩胶75 V和分离胶120 V恒压电泳),转膜45 min至PVDF膜上。转膜后4℃一抗(β-actin兔抗鼠单克隆抗体购自美国Santa Cruz公司,GCDH兔抗鼠多克隆抗体购自武汉三鹰生物技术有限公司,以1:100稀释)孵育过夜,二抗(辣根过氧化物酶标记羊抗兔二抗购于北京中杉金桥生物技术有限公司,以1:3 000稀释)孵育1 h,ECL显色。UVP Labworks照相,Labworks 4.6软件定量分析光密度值。每组每次实验设3个平行样本,整个实验独立重复3次,检测结果以GCDH相对于β-actin相对表达量表示。
1.4 MTT检测细胞活性BRL细胞经慢病毒感染72 h后传代培养于96孔板,分别用普通培养基和含5 mmol/L赖氨酸(美国Sigma公司)的培养基培养24 h后行MTT检测。每孔细胞(90 μL培养基)加5 mg/mL MTT(美国Amresco公司)10 μL,37℃、5%CO2培养箱中孵育4 h后,换DMSO室温摇床震荡15 min。570 nm测量波长、630 nm参照波长测定各孔光密度值(OD值)。每组设3个复孔,整个实验独立重复6次。
1.5 Hoechst 33342检测细胞凋亡BRL细胞经慢病毒感染72 h后传代培养于96孔板,分别用普通培养基和含5 mmol/L赖氨酸的培养基培养24 h后行Hoechst 33342检测。37℃避光,终浓度10 μg/mL Hoechest 33342(美国Sigma公司)孵育细胞10 min。4%多聚甲醛、避光固定细胞10 min,PBS洗涤细胞后荧光显微镜下观察。每组设3个复孔,每孔随机选择10个视野拍摄,计算Hoechest 33342染色阳性核比例。整个实验独立重复6次。
1.6 Western blot检测Caspase3表达水平BRL细胞经慢病毒感染72 h后传代培养于6孔板,继续用含5 mmol/L赖氨酸的培养基培养24 h后提取蛋白,检测方法同1.3小节(Caspase3兔抗鼠多克隆抗体购自美国Santa Cruz公司,以1:500稀释)。每组每次实验设3个平行样本,整个实验独立重复3次,检测结果以Caspase3相对于β-actin相对表达量表示。
1.7 统计学分析运用SPSS 17.0统计软件包对数据行统计学分析。计量资料用均数±标准差(x±s)表示,两组间比较采用独立样本t检验;多组间比较采用单因素方差分析,组间两两比较采用SNK-q检验。P < 0.05为差异有统计学意义。
2 结果 2.1 BRL肝细胞培养与慢病毒感染MOI=20时,倒置显微镜下可见感染后BRL细胞与正常对照组BRL细胞均呈不规则多角形,相互连接成片,胞核折光性好。荧光显微镜下可见阴性对照组和GCDH沉默组90%以上BRL细胞呈绿色荧光,提示被慢病毒成功感染。见图 1。

|
图 1 慢病毒感染BRL细胞(×200) MOI=20时,各组细胞在光镜下观察均呈不规则多角形,相互连接成片,胞核折光性好。被慢病毒成功感染的细胞在荧光显微镜下呈绿色荧光,提示阴性对照组和GCDH沉默组感染效率高达90%以上,且细胞活性好。 |
2.2 慢病毒感染后BRL肝细胞GCDH蛋白表达水平检测
各组GCDH蛋白表达量比较差异有统计学意义(F=196.17,P < 0.01)。与正常对照组和阴性对照组比较,GCDH沉默组GCDH蛋白表达量显著下降(P < 0.01);而正常对照组与阴性对照组间比较,GCDH蛋白表达量差异无统计学意义(P > 0.05)。见图 2。

|
图 2 Western blot检测各组细胞GCDH蛋白表达水平 上图为各组细胞GCDH蛋白表达电泳图;下图为各组细胞GCDH蛋白水平比较统计图(n=3),a示与正常对照组比较,P < 0.01;b示与阴性对照组比较,P < 0.01。 |
2.3 GCDH基因沉默及高浓度赖氨酸对各组BRL肝细胞活性的影响
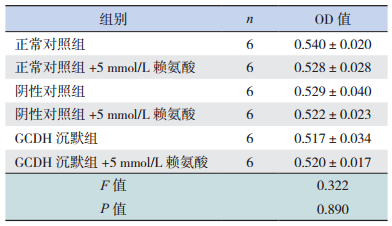
各处理组间OD值比较差异无统计学意义(P > 0.05),说明各处理组间细胞活性无明显差异,GCDH表达减少和5 mmol/L赖氨酸对BRL肝细胞活性均无影响,见表 1。
| 表 1 MTT检测各组BRL细胞OD值比较 (x±s) |
2.4 GCDH基因沉默及高浓度赖氨酸对各组BRL肝细胞凋亡的影响
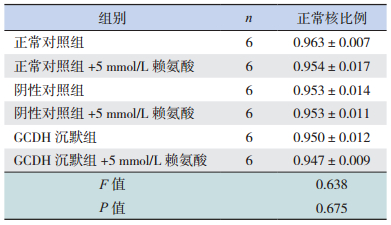
各组细胞胞核均被染成蓝色,未见明显局部深染高密度区或核碎片(图 3)。各组细胞正常细胞核比例均在95%以上,且各组间比较差异无统计学意义(P > 0.05),说明GCDH表达减少和5 mmol/L赖氨酸对BRL肝细胞核形态影响均不明显,见表 2。

|
图 3 各处理组细胞Hoechest 33342染色(荧光显微镜,×200) 各组细胞胞核均被染成蓝色,未见明显局部深染高密度区或核碎片。 |
| 表 2 Hoechest 33342染色检测各组BRL细胞正常核比例比较 (x±s) |
2.5 赖氨酸作用下GCDH沉默组与阴性对照组Caspase3表达水平
经5 mmol/L赖氨酸培养后,GCDH沉默组与阴性对照组间Caspase3表达水平比较差异无统计学意义(P > 0.05),说明GCDH表达减少和5 mmol/L赖氨酸对BRL肝细胞凋亡无影响。见图 4。

|
图 4 Western blot检测GCDH沉默组与阴性对照组间Caspase3表达水平变化 上图为电泳条带图;下图为统计图(n=3)。 |
3 讨论
GA1是一种常染色体隐性遗传疾病,是指编码GCDH的基因发生突变导致GCDH酶活性缺陷而引起的疾病。GCDH位于线粒体基质内,GCDH缺陷致色氨酸、赖氨酸、羟赖氨酸代谢障碍,戊二酸、3羟基戊二酸、戊二酰肉碱在体液和组织中浓度升高。GCDH广泛分布于脑、肾脏、肝脏、心脏、皮肤等多个器官[10]。肝脏作为氨基酸代谢和能量代谢的重要器官,急性脑病危象发作时常伴有低血糖、代谢性酸中毒、高氨血症等。GA1治疗指南中提出在感染、外伤等急性分解代谢状态时需及时补充葡萄糖等能量供给[5]。对GA1发病机制的探讨,肝脏是除脑之外的又一大重要器官。但GA1的临床表现主要为生后早期急性脑病危象后继发的以纹状体为主的神经系统损伤或年长儿甚至成人发病的隐匿性神经系统病变,少有肾脏损伤的报道,几乎未见肝脏的损伤[11]。高赖氨酸饮食饲养GCDH基因敲除小鼠模拟了GA1体内生化改变及纹状体为主的神经系统损伤,同时用该模型检测肝脏转氨酶并无改变[6]。GCDH基因敲除小鼠生后30 d时腹膜内注射赖氨酸致使其纹状体神经元出现明显的氧化应激损伤,而其肝脏和心脏并无明显损伤[7]。GCDH基因敲除小鼠生后15 d时腹膜内注射赖氨酸致使其肝脏及脑组织内还原性谷胱甘肽和巯基含量降低,羰基含量增加。但其脑组织的氧化应激损伤明显强于肝脏[8]。了解肝脏受损不明显的机制,有助于为GA1治疗提供新的理论依据。
前期研究证实携带靶向沉默GCDH基因的shRNA慢病毒感染原代培养纹状体神经元,结合高浓度赖氨酸(5 mmol/L)培养环境可有效模拟GA1内环境对纹状体神经元的损伤。并利用该细胞模型初步研究了GA1神经元损伤机制[9]。为了进一步研究GA1对肝脏的损伤作用,本研究用携带靶向沉默GCDH基因的shRNA慢病毒感染BRL肝细胞系,结合高浓度赖氨酸培养环境模拟GA1内环境对肝细胞活性的影响。慢病毒感染BRL肝细胞感染效率及GCDH蛋白沉默效率均增高。前期研究在GA1纹状体神经元模型中,阴性对照病毒及含5 mmol/L赖氨酸培养基对神经元细胞活性无影响;靶向沉默GCDH基因慢病毒促神经元凋亡,且5 mmol/L赖氨酸进一步促进该效应[9]。在该实验细胞模型中,MTT和Hoechst 33342染色实验,Caspase3水平检测均显示赖氨酸代谢累积物对肝细胞损伤不明显。
这些研究都提示在GA1患儿中脑组织,尤其是纹状体较周围其他器官组织更易出现损伤。据血液和尿液中戊二酸、3羟基戊二酸含量的高低,GA1可分为高分泌型和低分泌型2种生化表型。但是生化表型和临床表现间却无相关性,即体液中戊二酸、3羟基戊二酸浓度与神经系统损伤程度不相关[12]。CGDH是位于线粒体基质内的酶,赖氨酸从血液至胞内再至线粒体内代谢,同时其活性缺陷导致的戊二酸、3羟基戊二酸累积首先发生在线粒体内,进一步转运至胞浆外。代谢累积物胞内外的转运与ORC1/2、ODC、OGC等多种转运体蛋白有关[13]。这些转运体蛋白在GA1患儿神经组织和肝脏中含量可能不一致导致二者间损伤存在差异性。同时还有研究发现赖氨酸在肝脏和脑组织中的代谢存在差异[14]。在肝脏主要经线粒体酵母氨酸途径代谢,在脑组织主要经过氧化物酶体甲基哌啶途径代谢,二者最终的代谢产物都汇聚至线粒体中经GCDH代谢。代谢途径下游GCDH活性缺陷,上游代谢累积物及累积方式不同也可能是GA1患儿脑组织和肝脏组织损伤差异性的原因。
本研究构建含shRNA慢病毒载体靶向沉默GCDH基因,慢病毒感染BRL肝细胞系,结合高浓度赖氨酸培养环境建立GA1肝细胞研究模型。将其和与之对应的神经元模型对比研究GA1患儿脑组织和肝脏组织损伤差异性的机制。有望为GA1治疗提供新的理论依据。
| [1] | Govender R, Mitha A, Mubaiwa L. A review of patients with glutaric aciduria type 1 at Inkosi Albert Luthuli Central Hospital, Durban, South Africa[J]. S Afr Med J, 2017, 107 (3): 201–204. DOI:10.7196/SAMJ.2017.v107i3.11332 |
| [2] | Tsai FC, Lee HJ, Wang AG, et al. Experiences during newborn screening for glutaric aciduria type 1:diagnosis, treatment, genotype, phenotype, and outcomes[J]. J Chin Med Assoc, 2017, 80 (4): 253–261. DOI:10.1016/j.jcma.2016.07.006 |
| [3] | Yang L, Yin H, Yang R, et al. Diagnosis, treatment and outcome of glutaric aciduria type Ⅰ in Zhejiang Province, China[J]. Med Sci Monit, 2011, 17 (7): 55–59. |
| [4] | Jafari P, Braissant O, Bonafé L, et al. The unsolved puzzle of neuropathogenesis in glutaric aciduria type Ⅰ[J]. Mol Genet Metab, 2011, 104 (4): 425–437. DOI:10.1016/j.ymgme.2011.08.027 |
| [5] | Boy N, Mühlhausen C, Maier EM, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type Ⅰ:second revision[J]. J Inherit Metab Dis, 2017, 40 (1): 75–101. DOI:10.1007/s10545-016-9999-9 |
| [6] | Zinnanti WJ, Lazovic J, Wolpert EB, et al. A diet-induced mouse model for glutaric aciduria type Ⅰ[J]. Brain, 2006, 129 (Pt 4): 899–910. |
| [7] | Seminotti B, da Rosa MS, Fernandes CG, et al. Induction of oxidative stress in brain of glutaryl-CoA dehydrogenase deficient mice by acute lysine administration[J]. Mol Genet Metab, 2012, 106 (1): 31–38. DOI:10.1016/j.ymgme.2012.03.002 |
| [8] | Seminotti B, Ribeiro RT, Amaral AU, et al. Acute lysine overload provokes protein oxidative damage and reduction of antioxidant defenses in the brain of infant glutaryl-CoA dehydrogenase deficient mice:a role for oxidative stress in GA I neuropathology[J]. J Neurol Sci, 2014, 344 (1-2): 105–113. DOI:10.1016/j.jns.2014.06.034 |
| [9] | Gao J, Zhang C, Fu X, et al. Effects of targeted suppression of glutaryl-CoA dehydrogenase by lentivirus-mediated shRNA and excessive intake of lysine on apoptosis in rat striatal neurons[J]. PLoS One, 2013, 8 (5): e63084. DOI:10.1371/journal.pone.0063084 |
| [10] | Braissant O, Jafari P, Remacle N, et al. Immunolocalization of glutaryl-CoA dehydrogenase (GCDH) in adult and embryonic rat brain and peripheral tissues[J]. Neuroscience, 2017, 343 : 355–363. DOI:10.1016/j.neuroscience.2016.10.049 |
| [11] | Kölker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2:the evolving clinical phenotype[J]. J Inherit Metab Dis, 2015, 38 (6): 1059–1074. DOI:10.1007/s10545-015-9840-x |
| [12] | Wang Q, Li X, Ding Y, et al. Clinical and mutational spectra of 23 Chinese patients with glutaric aciduria type 1[J]. Brain Dev, 2014, 36 (9): 813–822. DOI:10.1016/j.braindev.2013.11.006 |
| [13] | Sauer SW. Biochemistry and bioenergetics of glutaryl-CoA dehydrogenase deficiency[J]. J Inherit Metab Dis, 2007, 30 (5): 673–680. DOI:10.1007/s10545-007-0678-8 |
| [14] | Sauer SW, Opp S, Hoffmann GF, et al. Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type Ⅰ[J]. Brain, 2011, 134 (Pt 1): 157–170. |