 2017, Vol. 19
2017, Vol. 19



2. 广州市妇女儿童医疗中心新生儿科, 广东 广州 510623;
3. 广东医科大学附属医院儿童医学中心, 广东 湛江 524001
微绒毛包涵体病(microvillus inclusion disease, MVID, OMIM #251850),又称为先天性微绒毛萎缩,是一种常染色体隐性遗传病,常表现为难治性水样腹泻、营养吸收障碍和生长发育迟缓[1-3]。MVID可分为早发型和迟发型,分别表现为出生后第1天和生后3~4月出现症状[4-5]。Davidson等[3]在1978年首次报道MVID,随后由Cutz等[1]命名。近年研究表明,90%的MVID由编码Vb型肌球蛋白的MYO5B基因突变导致,其余由编码突触融合蛋白的STX3基因突变导致[6]。目前国内外尚无关于本病的流行病学数据。我国学者虽有MYO5B基因突变与胆汁淤积或精神分裂有关的研究发现[7-8],但迄今国内文献中仅有1篇关于该基因突变导致MVID的论著[9]。本研究报道1例经遗传学分析确诊MVID患儿的诊治经过,为本病确诊提供遗传学依据,并为后续诊治研究提供参考。
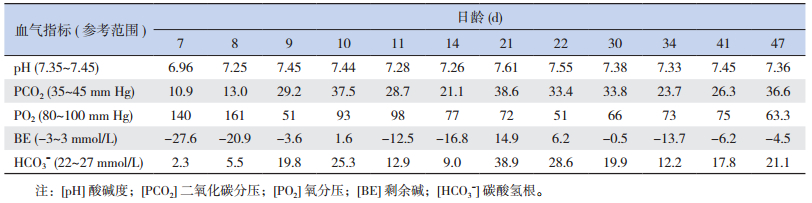
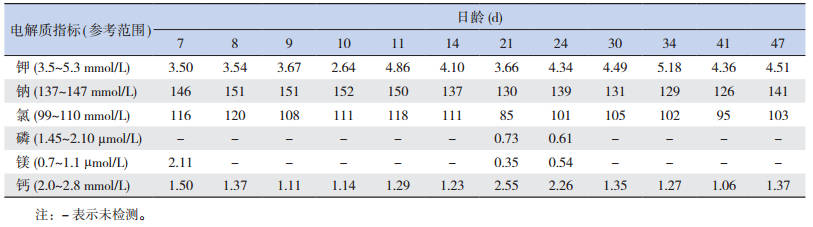
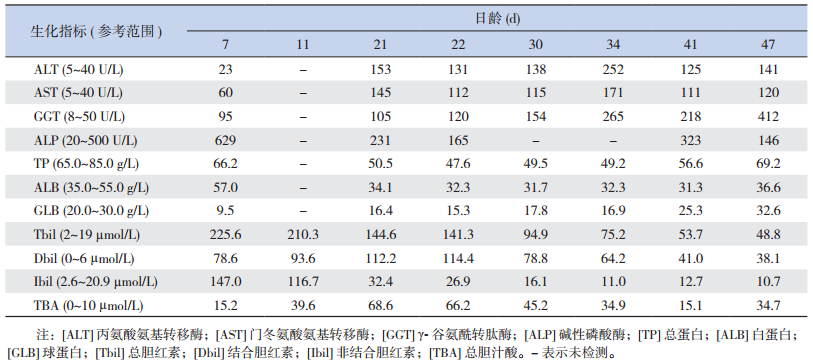
1 资料与方法 1.1 病例介绍患儿,女,21 d,因“解稀便20 d”入院。患儿生后2 d开始解稀便,7~10次/d,量少,伴腹胀;生后7 d仍有腹胀腹泻,并出现气促、精神反应差,遂至当地医院住院治疗。胃肠超声示:右侧腹部局部肠管稍扩张,腹腔肠管蠕动较快;血气分析示:代谢性酸中毒(表 1)、电解质紊乱(表 2);生化检查示:总胆红素、直接胆红素和胆汁酸等指标升高,提示胆汁淤积性黄疸(表 3);尿常规示:尿蛋白+、白细胞+、亚硝酸盐+、葡萄糖2+、酮体+、尿胆红素2+、潜血+、尿酸碱度6.0、尿比重1.020;血遗传代谢病筛查结果示:多种氨基酸增高。期间给予蒙脱石散止泻,枯草杆菌二联活菌和布拉氏酵母菌调节肠道功能,熊去氧胆酸利胆,更换无乳糖强化中链脂肪酸奶粉喂养,调节电解质平衡及静脉营养等治疗,共住院20 d,肝功能和酸中毒均未见明显改善。为明确病因遂至我科就诊。
| 表 1 患儿血气指标的动态改变 |
| 表 2 患儿历次电解质检查结果 |
| 表 3 患儿历次生化检查结果 |
患儿系第3胎第2产,出生胎龄39周,经阴道分娩出生,出生时羊水Ⅲ°混浊,无缺氧窒息史,1 min、5 min、10 min Apgar评分均为10分,脐带胎盘、胎膜无异常。出生体重2.85 kg。父母体健,非近亲结婚,否认家族类似病史及传染病史。
体格检查:体重2.2 kg(< -3 SD),身长45 cm(< -2 SD),头围33 cm。神志清醒,反应差,营养不良貌。皮肤、巩膜黄染,皮下脂肪2 mm,无皮疹及皮下出血点。前囟平软,大小约0.5 cm×0.5 cm。颈软,双锁骨触诊连续,双肺呼吸音清,未闻及干、湿性罗音。心律齐,心音有力,未闻及病理性杂音。腹软,脐部干洁,腹部膨隆,腹部静脉显露,最大腹围为33 cm,经脐腹围31 cm,腹部无包块。肝脾肋下未触及。移动性浊音(-),肠鸣音正常。脊柱、四肢无畸形,肛门及外生殖器无异常。腹壁、膝腱和跟腱等生理反射可引出,克氏、布氏和巴氏征均阴性。
辅助检查:入院后查血常规大致正常。生化检查发现电解质紊乱(表 2),同时胆汁酸、胆红素、转氨酶、谷氨酰转肽酶等胆汁淤积指标均升高(表 3)。血清锌5.70 µmol/L(参考值11.47~25.50 µmol/L)。
1.2 二代测序技术采集患儿及其父母的EDTA抗凝外周血2 mL,按照Blood DNA Mini kit试剂盒(杭州新景生物试剂开发有限公司)说明书提取基因组DNA。将基因组DNA打断,建立含有与消化系统相关基因(NEUROG3、SLC26A3、EPCAM、CTLA4、MYO9B、BSND、APC和MYO5B等共54个基因)的全基因组文库,并对文库样本进行定量,保证文库样本总量在3 µg以上。上述目标基因均用液相捕获试剂盒(北京迈基诺基因科技股份有限公司)捕获,利用新一代测序仪IlluminaHiSeq2000(美国Illumina公司)进行高通量测序。测序平均深度不小于200(乘数),获得原始数据,进行详细的基因序列生物信息学分析,找出相关基因的所有突变信息,同时判断是否具有致病性。
1.3 Sanger测序验证根据二代测序技术检测结果,对患儿及其父母基因组DNA进行Sanger测序,验证MYO5B基因突变。根据MYO5B基因序列(Ensemble Genome Browser: ENSG00000167306),运用Primer Premier 5.0软件设计引物(表 4),由北京迈基诺基因科技股份有限公司合成。聚合酶链反应体系包含2×Goldstar Buffer Mix(北京康为世纪生物科技有限公司)10 μL,上下游引物(10 μM)各1 μL,DNA 1 μL,加灭菌双蒸水7 μL至反应体系为20 μL。扩增条件为:95℃预变性10 min;随后分为4步,即第1步94℃变性30 s,64℃退火30 s,72℃延伸45 s,共反应3个循环;第2步94℃变性30 s,62℃退火30 s,72℃延伸45 s,共反应5个循环;第3步94℃变性30 s,60℃退火30 s,72℃延伸45 s,共反应10个循环;第4步94℃变性30 s,58℃退火30 s,72℃延伸45 s,共反应17个循环;最后再72℃延伸5 min。聚合酶链反应产物纯化后,由北京迈基诺基因科技股份有限公司进行Sanger测序。
| 表 4 MYO5B基因突变位点的Sanger测序引物 |

1.4 生物信息学分析
应用Chromas和DNAMAN软件对测序结果进行分析,并采用Human Splicing Finder(http://www.umd.be/HSF3/)、MaxEntScan(http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html)、NNSplice(http://www.fruitfly.org/seq_tools/splice.html)和NetGene2(http://www.cbs.dtu.dk/services/NetGene2)等在线软件工具,预测剪接位点变异是否影响前体mRNA剪接。
本研究经暨南大学附属第一医院医学伦理委员会批准,并请患儿父母签署书面知情同意书。
2 结果 2.1 遗传学分析结果二代测序技术分析消化系统相关的54个基因,在患儿MYO5B基因上检测到两个突变c.1966C > T(p.R656C)和c.310+2Tdup(图 1~2)。Sanger测序结果证实,患儿为上述突变的复合杂合子,而父母分别为以上突变的携带者(图 3)。前一个突变引起MYO5B蛋白(Vb型肌球蛋白)第656位氨基酸残基由精氨酸变为半胱氨酸,是国外文献已报道的致病性突变[10]。经检索中国知网、万方、维普和Pubmed等文献数据库,以及人类基因突变数据库(human gene mutation database, HGMD)和千人基因组计划人群数据库,证实c.310+2Tdup为未报道过的新突变。生物信息学预测结果提示,该突变对野生型供体位点具有强破坏性,同时产生了新的剪接供体位点(野生型CV值为33.94,突变型CV值为76.01,ΔCV值为+ 123.95%),从而影响前体mRNA剪接,阻碍MYO5B蛋白的正常合成。另外,MaxEntScan、NNSplice和NetGene2软件预测野生型剪接位点得分分别为10.65、1.00和0.99;而突变型剪接位点得分分别为-0.66、0.93和0.55;均提示该突变可能影响前体mRNA剪接。

|
图 1 MYO5B基因c.1966C > T突变的二代测序技术分析结果 箭头所指为MYO5B基因模板链第1966位点野生型鸟嘌呤G部分突变为腺嘌呤A(c.1966C > T)。 |

|
图 2 MYO5B基因c.310+2Tdup突变的二代测序分析结果 箭头所指为MYO5B基因模板链第310+2野生型腺嘌呤A部分重复(c.310+2Tdup)。 |

|
图 3 患儿及其父母MYO5B基因突变的Sanger测序验证结果 患儿为突变c.1966C > T和c.310+2Tdup的复合杂合子;父亲为c.310+2Tdup突变携带者;母亲为c.1966C > T突变携带者。 |
根据临床特点及遗传学分析结果,患儿最终确诊为MYO5B基因突变所致的MVID。
2.2 治疗与结局患儿以先天性腹泻、电解质紊乱、新生儿高胆红素血症、营养不良收住院。给予无乳糖强化中链脂肪酸奶粉喂养,以及胃肠减压、静脉营养等对症治疗,但疗效欠佳。患儿腹泻顽固,代谢性酸中毒(表 1)和电解质絮乱(表 2)难以纠正,且转氨酶、GGT、TBA、胆红素等胆汁淤积指标持续高于正常范围(表 3)。住院1月余自动出院,出院后失访。
3 讨论靶向Panel高通量测序作为二代测序技术的一种,能够针对性地检测出来源于同一通路结构的多种基因相关疾病,并对临床表现相同的遗传性疾病进行鉴别诊断;是一种低成本、有效、快速的分子诊断方法[10]。靶向Panel高通量测序与Sanger测序相结合,100%覆盖该Panel所含基因[11]。本研究患儿进行多次多项辅助检查均未找到病因,考虑为遗传性疾病;且仅靠临床特点难以确定致病基因,行二代测序技术分析消化系统疾病相关基因结合Sanger测序验证后,发现患儿为MYO5B基因突变c.310+2Tdup和c.1966C > T(p.R656C)的复合杂合子,其父母分别为以上突变的携带者,最终确诊MVID。
MYO5B基因位于染色体18q21.1,含有40个外显子和39个内含子,基因组全长372 kb,编码由1 848个氨基酸组成的Vb型肌球蛋白[4]。Vb型肌球蛋白可调节肠上皮细胞的膜运输和再循环[2];此外,MYO5B蛋白与Rab8a、Rab11a可形成Rab GTP酶-MYO5B复合物,不仅可以转运微绒毛的关键组成部分,促进微绒毛的生长,而且对顶端三磷酸腺苷结合盒转运蛋白(如BESP和MDR3)的转运功能和维持细胞极化有重要作用[12-16]。MYO5B基因突变所致的MVID中出现的难治性腹泻可能与肠上皮细胞顶端转运蛋白,如Rab8a、Rab11a和顶端转运钠氢交换体3的错位分布,致使肠上皮细胞的极化和刷状缘完整性受损有关[12, 17]。此外。MYO5B蛋白在肺、胃、肠和肾等多个组织的上皮细胞均表达[2];故MVID患者中常有肠外表现,如血尿、肾Fanconi综合征、低血磷性佝偻病、糖尿病、精神异常,以及心脏、肺和肝功能异常等[4]。本例患儿表现为严重而难治性的腹泻、胆汁淤积症、电解质絮乱、代谢性酸中毒,伴尿常规明显异常,符合MVID的临床表现。
2008年,Müller等[12]首次报道MYO5B基因突变与MVID有关。迄今为止,已发现40余种突变类型,包括错义、无义、缺失、插入、剪接位点突变等[3, 4, 7-9, 12, 18-20]。根据突变对Vb型肌球蛋白结构和功能的影响,可将其分为5种类型,包括与肌动蛋白相互作用的错义突变区、核苷酸结合的错义突变区、动力变构重排的错义突变区、蛋白错误折叠的突变区和蛋白质提前终止区[4]。本研究患儿的MYO5B基因突变c.1966C > T(p.R656C)位于动力变构重排的错义突变区,会影响Vb型肌球蛋白的变构运动重排,致使其动力功能受损[4, 12]。此外,经多个软件预测,剪接位点突变c.310+2Tdup可导致前体mRNA剪接异常,影响Vb型肌球蛋白的正常合成。两个突变共同作用,致使本研究患儿出现MVID的一系列临床表现。
大部分MVID患者常表现为以正常或低水平的血清GGT为特点的肝内胆汁淤积症,其发生机制可能与肝细胞的MYO5B/Rab11a顶端回收内体通路受损,毛细胆管膜上BSEP表达改变,以及回肠吸收胆汁酸和肝摄取胆汁酸增多有关[14]。此外,MVID患者因长期腹泻,吸收障碍常需要进行肠外营养,而肠外营养可引起胆汁淤积,表现为血清结合胆红素、碱性磷酸酶、转氨酶和GGT升高[21];故亦有小部分MVID患者中血清GGT呈高水平。本研究患者已进行肠外营养,且多次进行生化检查,发现刚开始GGT水平为轻度增高,后呈上升趋势。GGT水平的这种改变,考虑为上述因素的混合作用所致。而本研究患者肝内胆汁淤积进行性加重,其原因可能为:MVID患者的回肠胆汁酸吸收能力呈不同程度的受损[14];肠内胆汁酸浓度减低和毛细胆管膜分泌胆汁酸减少,可上调顶端钠依赖性胆汁酸转运蛋白,致使胆汁酸的肠重吸收增加,胆汁酸肝细胞毒性随之增加,加速肝脏疾病进展[21]。
MVID预后较差,目前尚无有效的药物治疗,常需行肠外营养和肠移植[4]。Ruemmele等[22]提出早期小肠移植结合结肠移植可明显改善MVID患者的预后,提高其生活质量。随后van Hoeve等[23]提出肠移植之前,进行不完全的小肠切除可改善水和电解质絮乱,从而提高生存率。另外,对于MYO5B基因突变造成胆汁淤积者,可给予熊去氧胆酸和部分胆汁分流术治疗,以改善症状[16]。本研究患儿给予无乳糖并强化中链脂肪酸奶粉喂养,以及胃肠减压、静脉营养和熊去氧胆酸治疗,疗效欠佳,且因患儿出院后失访,其远期预后无法随访观察。
综上所述,本研究报道了1例MVID患儿的临床表现和实验室检查特点。通过二代测序技术及Sanger测序验证,发现其为MYO5B基因突变c.310+2Tdup和c.1966C > T的复合杂合子,其中c.310+2Tdup为新的剪接位点突变,从而明确其病因诊断为MVID。本研究扩展了MYO5B基因突变谱,同时为患者分子诊断、临床管理及产前诊断提供了依据。
| [1] | Cutz E, Rhoads JM, Drumm B, et al. Microvillus inclusion disease:an inherited defect of brush-border assembly and differentiation[J]. N Engl J Med, 1989, 320 (10): 646–651. DOI:10.1056/NEJM198903093201006 |
| [2] | Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy)[J]. Orphanet J Rare Dis, 2006, 1 : 22. DOI:10.1186/1750-1172-1-22 |
| [3] | Davidson GP, Cutz E, Hamilton JR, et al. Familial enteropathy:a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy[J]. Gastroenterology, 1978, 75 (5): 783–790. |
| [4] | van der Velde KJ, Dhekne HS, Swertz MA, et al. An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations[J]. Hum Mutat, 2013, 34 (12): 1597–1605. DOI:10.1002/humu.22440 |
| [5] | Phillips AD, Schmitz J. Familial microvillous atrophy:a clinicopathological survey of 23 cases[J]. J Pediatr Gastroenterol Nutr, 1992, 14 (4): 380–396. DOI:10.1097/00005176-199205000-00003 |
| [6] | Michaux G, Massey-Harroche D, Nicolle O, et al. The localisation of the apical Par/Cdc42 polarity module is specifically affected in microvillus inclusion disease[J]. Biol Cell, 2016, 108 (1): 19–28. DOI:10.1111/boc.201500034 |
| [7] | Qiu YL, Gong JY, Feng JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis[J]. Hepatology, 2017, 65 (5): 1655–1669. DOI:10.1002/hep.29020 |
| [8] | Chen Y, Tian L, Zhang F, et al. Myosin Vb gene is associated with schizophrenia in Chinese Han population[J]. Psychiatry Res, 2013, 207 (1-2): 13–18. DOI:10.1016/j.psychres.2013.02.026 |
| [9] | 毛曼, 郭丽, 张占会, 等. 微绒毛包涵体病一家系的表型及遗传学分析[J]. 中华医学遗传学杂志, 2016, 33 (6): 792–796. |
| [10] | Xue Y, Ankala A, Wilcox WR, et al. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing:single-gene, gene panel, or exome/genome sequencing[J]. Genet Med, 2015, 17 (6): 444–451. DOI:10.1038/gim.2014.122 |
| [11] | Sun Y, Ruivenkamp CA, Hoffer MJ, et al. Next-generation diagnostics:gene panel, exome, or whole genome?[J]. Hum Mutat, 2015, 36 (6): 648–655. DOI:10.1002/humu.22783 |
| [12] | Müller T, Hess MW, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity[J]. Nat Genet, 2008, 40 (10): 1163–1165. DOI:10.1038/ng.225 |
| [13] | Engevik AC, Goldenring JR. Trafficking ion transporters to the apical membrane of polarized intestinal enterocytes[J]. Cold Spring Harb Perspect Biol, 2017.[Epub ahead of print] |
| [14] | Girard M, Lacaille F, Verkarre V, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease[J]. Hepatology, 2014, 60 (1): 301–310. DOI:10.1002/hep.v60.1 |
| [15] | Siahanidou T, Koutsounaki E, Skiathitou AV, et al. Extraintestinal manifestations in an infant with microvillus inclusion disease:complications or features of the disease?[J]. Eur J Pediatr, 2013, 172 (9): 1271–1275. DOI:10.1007/s00431-013-1948-0 |
| [16] | Gonzales E, Taylor SA, Davit-Spraul A, et al. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease[J]. Hepatology, 2017, 65 (1): 164–173. DOI:10.1002/hep.28779 |
| [17] | Vogel GF, Janecke AR, Krainer IM, et al. Abnormal Rab11-Rab8-vesicles cluster in enterocytes of patients with microvillus inclusion disease[J]. Traffic, 2017, 18 (7): 453–464. DOI:10.1111/tra.2017.18.issue-7 |
| [18] | Erickson RP, Larson-Thomé K, Valenzuela RK, et al. Navajo microvillous inclusion disease is due to a mutation in MYO5B[J]. Am J Med Genet A, 2008, 146A (24): 3117–3119. DOI:10.1002/ajmg.a.v146a:24 |
| [19] | Szperl AM, Golachowska MR, Bruinenberg M, et al. Functional characterization of mutations in the myosin Vb gene associated with microvillus inclusion disease[J]. J Pediatr Gastroenterol Nutr, 2011, 52 (3): 307–313. DOI:10.1097/MPG.0b013e3181eea177 |
| [20] | Ruemmele FM, Müller T, Schiefermeier N, et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease:15 novel mutations and a CaCo-2 RNAi cell model[J]. Hum Mutat, 2010, 31 (5): 544–551. DOI:10.1002/humu.v31:5 |
| [21] | Kelly DA. Preventing parenteral nutrition liver disease[J]. Early Hum Dev, 2010, 86 (11): 683–687. DOI:10.1016/j.earlhumdev.2010.08.012 |
| [22] | Ruemmele FM, Jan D, Lacaille F, et al. New perspectives for children with microvillous inclusion disease:early small bowel transplantation[J]. Transplantation, 2004, 77 (7): 1024–1028. DOI:10.1097/01.TP.0000119163.30745.C1 |
| [23] | van Hoeve K, Hoffman I, Fusaro F, et al. Microvillus inclusion disease:a subtotal enterectomy as a bridge to transplantation[J]. Acta Chir Belg, 2016, 116 (6): 333–339. DOI:10.1080/00015458.2016.1176420 |