PDF(1302 KB)
PDF(1302 KB)
 PDF(1302 KB)
PDF(1302 KB)
PDF(1302 KB)
PDF(1302 KB)
新生儿难治性惊厥STXBP1基因突变的研究
STXBP1 gene mutation in newborns with refractory seizures
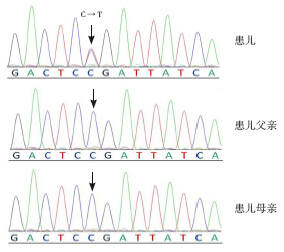
目的 探讨STXBP1 基因突变与不明原因新生儿难治性惊厥的关系。方法 应用直接测序法对11 例原因不明的新生儿难治性惊厥患儿进行STXBP1 编码区的突变检测。结果 11 例患儿中,1 例检测到STXBP1 基因突变。突变类型为错义突变,突变位点c.1439C>T(p.P480L)。结论 在新生儿难治性惊厥患儿中,可检测到STXBP1 基因突变,为该类疾病的深入研究提供了新的思路。
Objective To study the relationship between STXBP1 gene mutations and refractory seizures with unknown causes in newborns. Methods The coding region of STXBP1 gene was detected using direct Sanger sequencing in 11 newborns with refractory seizures of unknown causes. Results STXBP1 gene mutation was found in 1 out of 11 patients. It was a missense mutation: c.1439C>T (p.P480L). Conclusions STXBP1 gene mutation can be found in neonatal refractory seizures of unknown causes, suggesting a new approach of further research of this disease.